
Technical details
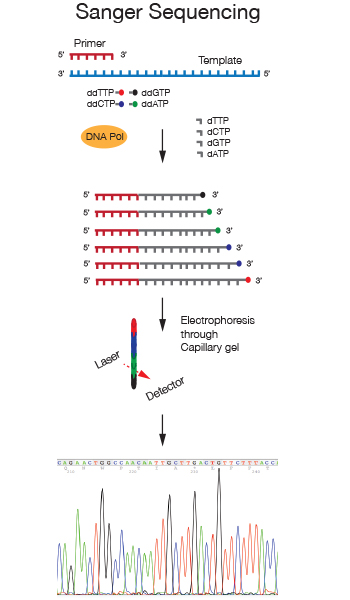
Sanger sequencing is a chain terminator sequencing method developed in the Seventies by Frederick Sanger. In this method the DNA strand analyzed is primed by an oligonucleotide and serves as a template for extension by a DNA polymerase. Chain termination is achieved through the addition of the nucleotide analog 2',3'-dideoxynucleotides (ddNTPs) to the nucleotide mix in the reaction. When a dideoxynucleotide is incorporated at the 3' end of the growing chain, chain elongation is terminated selectively at A, C, G, or T because the ddNTP lacks a 3'-hydroxyl group.
We use Dye terminator sequencing, in which four differently fluorescently labeled ddNTPs allow detection of the four bases due to different emission spectra. After separation of the sequencing reaction on capillary gels, the differently labeled fragments are detected and the 4 nucleotides in the sequence are displayed as differently colored peaks in a chromatograph.
If you have many samples that need to be sequenced with the same primer, it is more convenient to provide the samples in a 96-well plate format. This way you can use the dedicated request form, which is quicker to fill in, we are quicker in pipetting and as a consequence the price/sample is reduced.
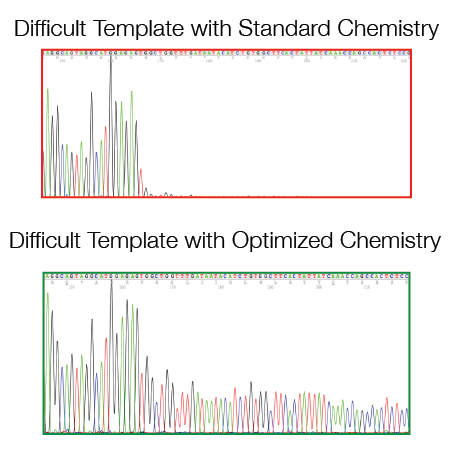
There are some templates/regions that are difficult to sequence when using a standard DNA sequencing protocol:
- GC rich regions (> 60% GC)
- Di- and Tri-nucleotide repeats
- Hairpin structures (e.g. si/shRNA inserts and pDONR/pDEST vector series from Invitrogen)
- A/T homopolymers (e.g. polyA resulting from oligodT primed amplification of mRNAs).
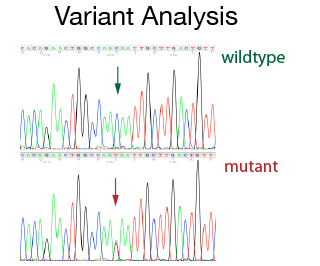
For many cases single strand sequencing gives you enough information. In specific cases where you need to be a 100% sure about the result, a more accurate result can be obtained by sequencing both strands of the DNA. This double strand strategy is required for variant detections (e.g. SNP or mutations) both at diagnostic and at a research level. The double strand sequencing strategy is also used for publication grade data or for specific projects (e.g. primer walking). The DNA Sequencing Staff will help with primer design and with the set up of the PCR conditions. In addition, they will do the data assembly and analysis.
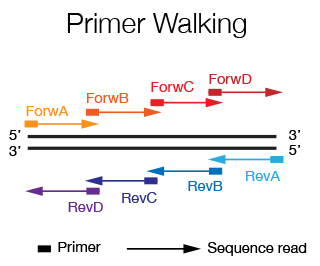
When you want to sequence an insert that is more than 1.2 Kb long, it is necessary to use the primer walking strategy. Starting by sequencing with primers that flank the insert, new primers are designed at the end of the sequence that is obtained until the entire sequence of the insert is covered. If the sequence is known, the primers can by designed a priori and are spaced every 500-600 bp. The DNA Sequencing Staff will help you in primer design and in data alignment/editing if needed.
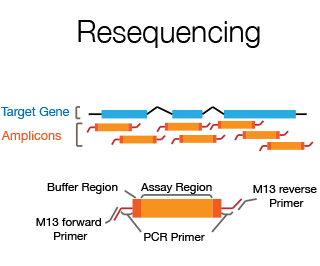
Resequencing is the sequencing of part of an individual's genome in order to detect sequence differences between the individual and the reference genome. Usually it involves sequencing of exons, splice junctions and UTRs of a specific candidate disease gene using the double strand sequencing approach. The DNA Sequencing Staff will help with primer design, set up of PCR conditions and do the data assembly and analysis.
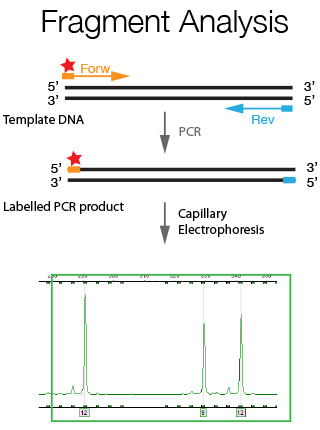
Fragment analysis is a general term used to describe experiments, which rely on the detection of changes in the length of a specific DNA sequence to indicate the presence or absence of a genetic marker. For this purpose the genomic region of interest is amplified using fluorescently labeled primers and the generated PCR fragments are resolved through a capillary sequencer. A resolution of size difference of up to 1 bp can be achieved with this technique. Fragment Analysis has a wide array of applications such as Microsatellite Analysis, Cell line verification and CrispR/Cas9 genotyping as further explained below.
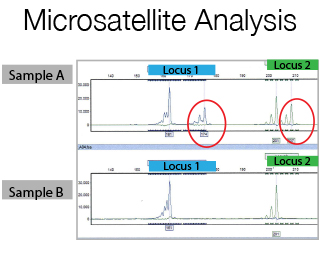
Microsatellites are di-, tri-, or tetra nucleotide tandem repeats in DNA sequences. The number of repeats is variable among different persons and within the alleles of an individual. If you flank a microsatellite with fluorescent PCR primers and amplify it, you will have a pair of fluorescent allelic products that will vary in size according to their repeat length. For Microsatellite studies a prior meeting with the Staff is required for primer design and experimental set-up.
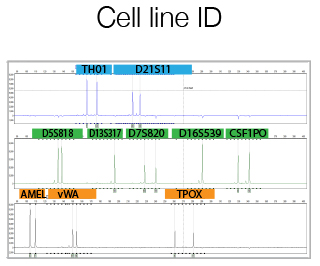
For human cell line the GenePrint® 10 System by Promega is used. This kit allows co-amplification and three-color detection of ten human loci (STR regions). These loci collectively provide a genetic profile with a random match probability of 1 in 2.92 × 109. An internal lane standard (ILS) and allelic ladder are provided for sizing and genotyping of amplified fragments, and a control DNA is supplied as a positive control. An optimized protocol is supplied by the Staff to help you using the Promega kit. The Service offers, in addition to the run of the reactions, also the comparison between the results and specific DataBase informations, previously provided by the user.
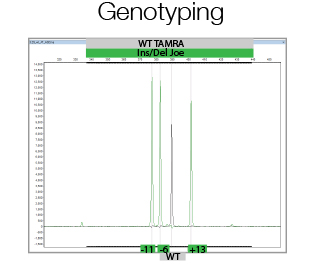
In its most simple form, CrispR/Cas9 genome editing creates double strand breaks, which in the majority of the cases are repaired using Non Homologous End Joining thereby creating small Insertions or Deletions. This is used to generate out-of-frame mutations in the gene of interest to create functional knock-outs. Due to the generation of Insertions/Deletions during this process, Fragment Analysis can be used to analyze the efficiency of genome editing of bulk populations of cells as well as to assess the allele status of single clones. The facility will assist in experimental and primer design, and in data analysis.
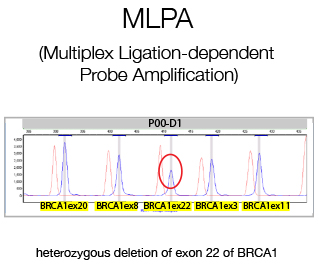
MLPA (Multiple Ligation Probe Amplification) is a multiplex PCR method detecting abnormal copy numbers and in clinical settings is used for the detection of genetic disorders caused by large rearrangements. We perform diagnostic MLPA for the Cancer Genetic Testing Laboratory for all patients lacking pathological variants as determined by Sanger Sequencing.
More informations
- Sanger Sequencing of DNA (Part I)
- Sanger Sequencing of DNA (Part II)
- Isolating Plasmid DNA [great 10 min video on plasmids and miniprep (1st year students). Start at 2.52 minutes if you want to go directly to the miniprep (uses Qiagen kit)].
