
Test genetici
Cogentech offre test genetici per la valutazione del rischio di cancro utilizzando due pannelli proprietari con tecnologia Next Generation Sequencig (NGS): OncoPan®, per le più comuni sindromi tumorali ereditarie dell'adulto, e OncoPed®, per le sindromi tumorali ereditarie pediatriche e rare.
OncoPan® valuta non solo la presenza di varianti patogenetiche in 45 geni predisponenti al cancro, ma anche 313 SNP per il calcolo del punteggio di rischio poligenico applicato al tumore della mammella e delle ovaie.
Il test di accertamento delle varianti nei familiari, per identificare i potenziali portatori, viene eseguito utilizzando il sequenziamento Sanger o l'analisi MLPA.
I test vengono generalmente eseguiti su campioni di sangue, ma possono essere effettuati anche su tamponi buccali.
Medicina personalizzata
Il pannello OncoPan®, oltre a identificare varianti che aumentano il rischio di cancro, è anche progettato per aiutare a indirizzare la terapia con gli inibitori di PARP (PARPi):
- Test somatico BRCA1/2 per la terapia con PARPi nel cancro al seno, alle ovaie, alla prostata e al pancreas. Questo test viene offerto a scopo di ricerca dal laboratorio di Milano.
- Test somatico OncoHRD per la terapia con PARPi nel carcinoma ovarico. Questo test viene offerto a scopo di ricerca dal laboratorio di Milano.
I test vengono generalmente eseguiti su campioni tumorali FFPE.

Test dei tumori ereditari
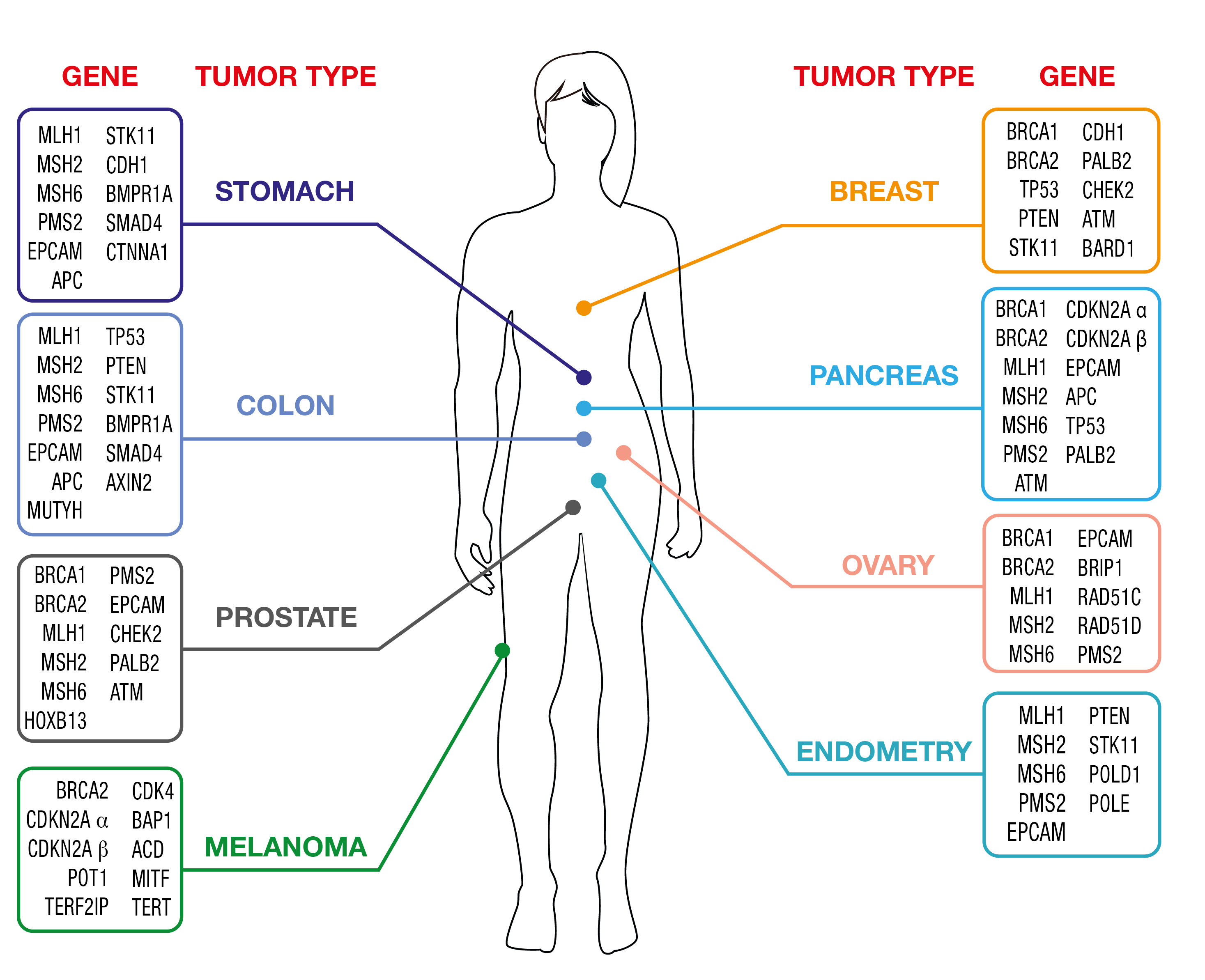
OncoPan® Sindromi tumorali ereditarie dell'adulto
La diagnostica del cancro è un campo in rapida evoluzione e i tumori ereditari non fanno eccezione. Oncopan risponde alle esigenze in rapida evoluzione essendo un pannello NGS innovativo e modulare che copre 45 geni e 313 SNP aggiuntivi per il calcolo di un punteggio di rischio poligenico (PRS). I medici possono richiedere pannelli ristretti per i test genetici, che possono essere integrati dall'analisi di ulteriori geni inclusi in Oncopan® se l'analisi iniziale non fosse informativa. Questo approccio riduce i costi e i tempi di diagnosi, migliorando al contempo i tassi di individuazione. Inoltre, il punteggio PRS sarà reso disponibile e potrà essere utilizzato dal medico per ottimizzare la determinazione del rischio di cancro utilizzando strumenti web dedicati (www.canrisk.org).
In base alla frequenza con cui queste sindromi si manifestano nella popolazione generale, OncoPan®viene utilizzato principalmente per i test genetici sul cancro ereditario della mammella e dell'ovaio (HBOC) e sulle sindromi del cancro colorettale
Clicca qui per un elenco delle sindromi tumorali ereditarie e dei geni associati inclusi in OncoPan®
Clicca qui per la scheda tecnica di OncoPan®.
Le donne portatrici di varianti patogene nei geni BRCA1 o BRCA2 hanno un rischio complessivo di circa il 70% di sviluppare un tumore al seno nel corso della vita, un rischio maggiore di tumore al seno controlaterale e di tumore alle ovaie. I portatori maschi sono particolarmente esposti al cancro alla prostata, con un certo rischio di cancro al seno maschile. Le prove scientifiche dimostrano anche un aumento del rischio per altri tipi di neoplasie, come il melanoma, il cancro allo stomaco, al pancreas e ai dotti biliari.
| Cancro ereditario associato a mutazioni in BRCA1/2 | % di rischio associato a | |
|---|---|---|
| BRCA1 | BRCA2 | |
| Cancro al seno | 72 | 69 |
| Tumore al seno controlaterale | 40 | 26 |
| Cancro ovarico | 44 | 17 |
| Cancro al seno maschile | 1-5 | 5-10 |
| Cancro alla prostata | - | 27 |
Una piccola percentuale di tumori al seno ereditari è legata alla presenza di varianti in altri geni, come PALB2, TP53, PTEN, STK11, CDH1. Per questi geni, il rischio di sviluppare un tumore al seno è ancora elevato e varia dal 35% al 60%, con variazioni da gene a gene. Alcuni di essi sono associati a specifiche sindromi genetiche che predispongono a una varietà di neoplasie ben definite.
Un aumento del rischio di tumore ovarico, circa il 15%, può essere dovuto alla presenza di varianti in geni come RAD51C, RAD51D, BRIP1. Inoltre, le variazioni nei geni della riparazione dei disallineamenti (MMR), cioè MLH1, MSH2, MSH6, PMS2 e le grandi delezioni del gene EPCAM, che predispongono all'insorgenza di tumori del colon associati alla sindrome di Lynch, possono conferire suscettibilità al carcinoma ovarico con rischi associati del 10%-17%.
Infine, sono stati identificati geni che conferiscono un rischio moderato-moderato di sviluppare un tumore al seno (ATM, CHEK2, BARD1) e diverse centinaia di variazioni nucleotidiche (SNPs) il cui effetto combinato (Polygenic risk score, PRS) può modulare il rischio conferito dai principali geni di suscettibilità BRCA1, BRCA2 , PALB2, ATM e CHEK2.
Negli ultimi anni i test per il cancro ereditario della mammella e dell'ovaio (HBOC) sono aumentati grazie al fatto che un maggior numero di ospedali ha iniziato a fornire il test ai propri pazienti.
Inoltre, è evidente la tendenza a sostituire i test per il solo BRCA1/2 con pannelli di test più ampi per consentire una migliore diagnosi. Ciò è dovuto al fatto che, sebbene queste forme di predisposizione abbiano generalmente manifestazioni cliniche specifiche, in alcuni casi non sono ben definite e possono in qualche misura sovrapporsi. L'introduzione di pannelli multigenici in Next Generation Sequencing (NGS) come OncoPan® consente un approccio "one-step", particolarmente utile nei casi di eterogeneità genetica.
| HBOC testing | 2020 | 2021 | 2022 | 2023 | 2024 | 2025 |
|---|---|---|---|---|---|---|
| BRCA | 445 | 313 | 290 | 270 | 217 | 114 |
| BRCA1/2 + PALB2 | 89 | 227 | 301 | 332 | 382 | 483 |
| > Pannello di 10 geni per HBOC | 14 | 170 | 445 | 727 | 1033 | 1004 |
| TOTAL HBOC TESTING | 548 | 710 | 1036 | 1329 | 1632 | 1601 |
Scelta del pannello per i test HBOC durante l'ultima triennale. La tendenza è quella di eseguire i test con pannelli che contengono più di 10 geni.
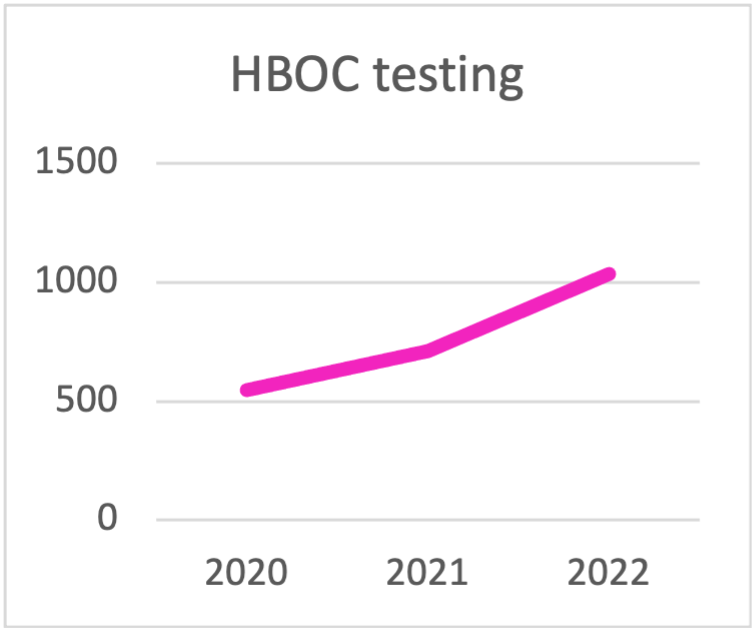
Test per HBOC eseguiti da Cogentech nell'ultima triennale
OncoPan® ha incorporato 313 SNP (polimorfismo a singolo nucleotide) per il calcolo del PRS Z-score con un algoritmo dedicato che Cogentech ha implementato. I punteggi di rischio poligenico sono una componente importante della previsione del rischio di cancro al seno e spiegano oltre il 30% del cancro al seno ereditabile. I punteggi di rischio poligenico si basano sul risultato combinato di varianti genetiche comuni. Con un software dedicato (www.canrisk.org), un medico professionista può utilizzare il PRS per calcolare meglio il rischio di sviluppare il cancro al seno e alle ovaie nelle donne, integrando anche altri fattori di rischio come lo stile di vita, i risultati dei test genetici e la storia familiare. Attualmente, il PRS per il cancro al seno e alle ovaie è stato convalidato solo in coorti europee e sono necessari e in corso ulteriori test per le diverse ascendenze.
Le più comuni predisposizioni ereditarie a sviluppare tumori del colon-retto sono la sindrome di Lynch, causata da alterazioni nei geni MLH1, MSH2, MSH6, PMS2, EPCAM, e la poliposi familiare, causata prevalentemente da mutazioni nei geni APC e MUTYH. Inoltre, i geni SMAD4 e BMPR1A sono associati all'insorgenza della poliposi giovanile. Recentemente sono stati identificati nuovi geni di predisposizione alla poliposi e ai tumori del colon: POLE e POLD1 (poliposi associata a mutazioni nei domini esonucleasici delle DNA polimerasi), NTHL1, MSH3 e AXIN2. Forme di polipi e adenomi del colon sono presenti anche in pazienti con la sindrome di Cowden, causata da varianti del gene PTEN, o con la sindrome di Peutz-Jeghers, causata da varianti del gene STK11. I tumori gastrici di tipo diffuso possono essere dovuti a mutazioni germinali nel gene CDH1. Recentemente, è stato identificato un nuovo gene di predisposizione, CTNNA1, che finora è stato segnalato in pochi casi, ma che dovrebbe essere considerato per la valutazione del rischio.
Queste forme di predisposizione hanno generalmente manifestazioni cliniche specifiche. Tuttavia, in alcuni casi, non sono ben definite e possono in qualche misura sovrapporsi. In effetti, sulla base dei dati della letteratura, è sempre più evidente che analizzare solo i geni più indicativi per ciascuna condizione può essere limitante. L'introduzione di pannelli multigenici nel sequenziamento di nuova generazione (NGS) consente un approccio "one-step", particolarmente utile nei casi di eterogeneità genetica. Questo approccio riduce i costi e i tempi di diagnosi, migliorando al contempo i tassi di individuazione. In conclusione, l'analisi completa che utilizza pannelli multi-gene può fornire risultati più informativi in termini di percentuale di pazienti idonei alla sorveglianza o alla terapia. Per questo motivo sono ampiamente utilizzati in ambito diagnostico.
| 2020 | 2021 | 2022 | 2023 | 2024 | 2025 | |
|---|---|---|---|---|---|---|
| Lynch | 123 | 131 | 113 | 160 | 138 | 197 |
| FAP | 42 | 30 | 22 | 9 | 21 | 16 |
| > Pannello di 10 geni per il colon | 19 | 25 | 47 | 51 | 78 | 89 |
| TOTAL TESTING | 184 | 186 | 182 | 220 | 237 | 302 |
Scelta del pannello per i test sulla sindrome del cancro ereditario del colon durante l'ultima triennale. La tendenza è quella di eseguire i test con pannelli che contengono più di 10 geni.
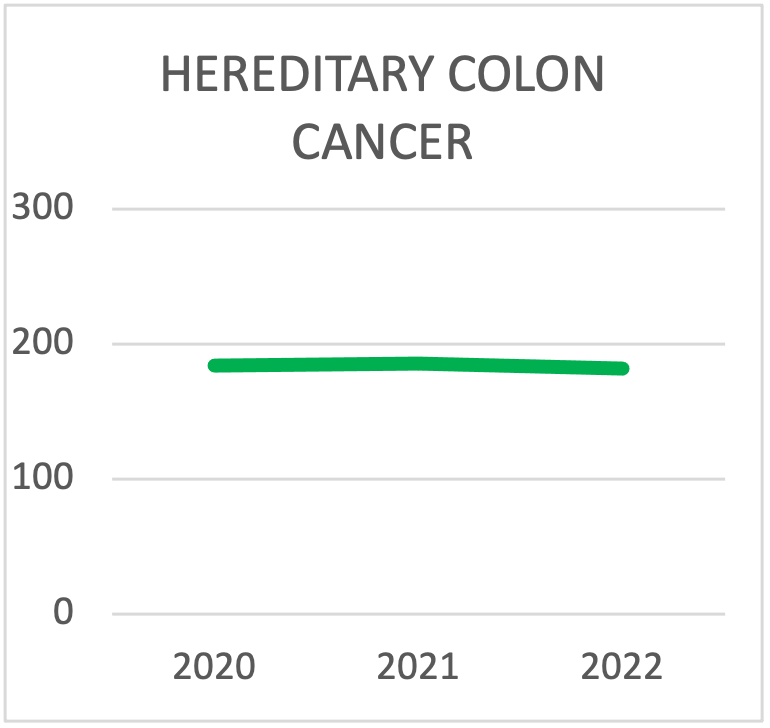
Test per le sindromi tumorali gastrointestinali eseguiti da Cogentech nell'ultimo triennio
La sindrome di Lynch è una sindrome oncologica autosomica dominante associata a una predisposizione genetica a diversi tipi di cancro. È nota anche come cancro colorettale ereditario non poliposico (HNPCC). La sindrome di Lynch è una delle sindromi tumorali ereditarie più comuni, con una prevalenza stimata dello 0,3% nella popolazione generale. La diagnosi di tumori del colon-retto, dell'endometrio, delle ovaie e/o di altri tipi di tumori in più parenti dello stesso lato della famiglia e la giovane età della diagnosi possono indicare la sindrome di Lynch. Le persone affette dalla sindrome di Lynch possono sviluppare più tipi di cancro nel corso della loro vita. Attualmente, i test per la sindrome di Lynch riguardano i geni MLH1, MSH2, MSH6, PMS2 e EPCAM.
La poliposi adenomatosa familiare (FAP) è una malattia autosomica dominante che consiste nello sviluppo di centinaia o addirittura migliaia di polipi nella mucosa del colon già a partire dalla pubertà. I polipi sono presenti nel 50% dei pazienti all'età di 15 anni e nel 95% all'età di 35 anni. Il cancro si sviluppa prima dei 40 anni in quasi tutti i pazienti non trattati. I pazienti possono anche sviluppare manifestazioni extracoloniche, come tumori desmoidi, osteomi del cranio o della mascella, cisti sebacee e adenomi in altre parti del tratto gastrointestinale. I pazienti hanno un rischio maggiore di cancro al duodeno, al pancreas, alla tiroide, di medulloblastomi e di epatoblastoma nei bambini. Nella maggior parte dei casi la FAP è legata alla mutazione germinale del gene APC.
La poliposi associata a MUTYH (MAP) è una malattia autosomica recessiva che si sviluppa in età adulta. La MAP si presenta spesso con un fenotipo attenuato, cioè con lo sviluppo di poche decine di polipi colorettali, che possono evolvere in carcinomi del colon, ed è dovuta a mutazioni bialleliche nel gene MUTYH.
La sindrome di Li-Fraumeni (LFS) è una sindrome ereditaria di predisposizione al cancro causata da una mutazione in p53, un gene soppressore del tumore. La sindrome prende il nome dai dottori Li e Fraumeni, che la descrissero per la prima volta nel 1969. I pazienti affetti tendono ad essere giovani, con la metà degli individui colpiti che riceve la prima diagnosi prima dei 30 anni. I tumori più comuni che si sviluppano nei pazienti affetti sono l'osteosarcoma, il sarcoma dei tessuti molli, la leucemia acuta, il cancro al seno, il cancro al cervello, i tumori della corticale surrenale e, meno frequentemente, una serie di altri tumori. Il rischio di sviluppare un tumore nel corso della vita per una persona affetta da LFS è del 90%, per questo è importante disporre di un protocollo di screening efficace.
La sindrome di Cowden (CS) fa parte della sindrome del tumore amartomatoso causata da mutazioni in PTEN. La sindrome di Cowden è una rara condizione autosomica dominante che comporta per le pazienti di sesso femminile un rischio del 50-85% di sviluppare un tumore al seno nel corso della vita. Altri tipi di cancro comportano un rischio minore, come il cancro della tiroide (30-40%), il cancro del rene (30-35%), il cancro dell'endometrio (25-30%), il cancro del colon-retto (5-10%) e il melanoma (6%).
La sindrome di Peutz-Jeghers (PJS) è causata da mutazioni in STK11, un gene soppressore del tumore. La sindrome di Peutz-Jeghers è un raro disturbo autosomico dominante che causa la crescita di polipi amartomatosi nel tratto digestivo nei pazienti affetti, generalmente entro i 10 anni di età. Le complicazioni di queste estese escrescenze benigne possono essere emorragie o blocchi intestinali. Il rischio che una persona affetta da PJS sviluppi un tumore nel corso della vita è del 93%. Il rischio più elevato è quello del cancro al seno (30-50%), seguito da quello del colon-retto (40%), del pancreas (10-35%), dello stomaco (30%), delle ovaie (20%), dei polmoni (15%) e da una serie di altri tumori a rischio minore.
Il cancro gastrico diffuso ereditario (HDGC) è una rara condizione ereditaria in cui gli individui hanno un rischio maggiore di sviluppare un cancro gastrico diffuso e un cancro al seno lobulare. Circa il 20% di tutti i tumori dello stomaco sono tumori gastrici diffusi, e un piccolo numero di questi è dovuto all'HDGC. I tumori dello stomaco sono definiti diffusi quando coinvolgono la maggior parte dello stomaco, causando un ispessimento della parete dello stomaco invece di rimanere in un'unica area e formare una massa distinta. Il gene coinvolto nell'HDGC è il CDH1, anche se in alcuni casi è stato dimostrato che anche il CTNNA1 è mutato.
Il 5-12% di tutti i casi di melanoma cutaneo appartengono a famiglie predisposte al melanoma o a famiglie con cluster multicancerosi correlati al melanoma, causati da alleli ad alta e media penetranza in geni di predisposizione noti come CDKN2A, CDK4, POT1, BAP1, TERT, ACD, TERF2IP, MITF e ATM. CDKN2A e CDK4 sono geni ad alta penetranza associati a un aumento del rischio di melanoma familiare. La prevalenza di varianti patogene o probabilmente patogene nel gene CDKN2A è di circa il 33% e fino al 58% nei casi di melanoma primario multiplo.
La sindrome di predisposizione ai tumori BAP1 è un disturbo ereditario che aumenta il rischio di una varietà di tumori benigni e cancerosi, più comunemente alcuni tipi di tumori che si verificano nella pelle, negli occhi, nei reni e nel mesotelio. Gli individui affetti possono sviluppare uno o più tipi di tumori e i membri affetti della stessa famiglia possono presentare tipi diversi. La BAP1-TPDS è causata da mutazioni germinali nel gene BAP1. I tumori più comuni sono il melanoma uveale, il mesotelioma maligno e il carcinoma renale a cellule chiare.
ONCOPED® - TUMORI PEDIATRICI E SINDROMI TUMORALI GENETICHE RARE
OncoPed® è indicato per l'analisi dei geni che conferiscono suscettibilità ai tumori che si sviluppano prevalentemente in età pediatrica e per la valutazione di rare condizioni sindromiche che possono svilupparsi anche in età adulta (ad esempio, melanomi e neoplasie renali).
La causa alla base della maggior parte dei tumori pediatrici è attualmente sconosciuta. Tuttavia, è stato stimato che le alterazioni genetiche, responsabili di una maggiore suscettibilità allo sviluppo del tumore, sono presenti in circa l'8% dei pazienti pediatrici, anche in assenza di una storia familiare significativa.
OncoPed® fornisce l'analisi dei geni coinvolti nei tumori pediatrici più aggressivi come il medulloblastoma, il tumore di Wilms, alcuni rari tumori ovarici (Sertoly-Leydig e SCCOTH), il retinoblastoma, il melanoma/melanoma uveale, i tumori renali, alcuni linfomi, i tumori rabdoidi e i tumori che si sviluppano nelle sindromi di Li Fraumeni, Cowden, Gorlin, DICER1 e Carney. Alcuni dei geni presenti in Oncopan® sono presenti anche in OncoPed®, sia perché la sindrome in questione si presenta sia nei giovani che negli adulti, sia perché il fenotipo del tumore si presenta in modo eterogeneo. Le sindromi per le quali il test è unico con OncoPed® sono elencate di seguito.
Clicca qui per un elenco delle sindromi tumorali ereditarie e dei geni associati inclusi in OncoPed®.
Clicca qui per la scheda tecnica di OncoPed®.
Le mutazioni bialleliche nei geni MLH1, MSH2, MSH6, PMS2 ed EPCAM, che sono coinvolti nella sindrome di Lynch, possono causare la sindrome da deficit costituzionale di riparazione degli accoppiamenti (CMMRD). La CMMRD è una rara malattia ereditaria che predispone a tumori maligni con esordio nell'infanzia, di solito cancro al cervello, ematologico e gastrointestinale.
La sindrome del carcinoma basocellulare nevoide (NBCCS), nota anche come sindrome di Gorlin, è una malattia ereditaria multisistemica che causa anomalie dello sviluppo e predisposizione a determinati tumori come il carcinoma basocellulare (BCC) e, più raramente, i medulloblastomi infantili. Fino all'85% dei casi è dovuto a mutazioni (varianti patogene) nel gene PTCH1. Più recentemente, un fenotipo simile è stato riscontrato in associazione a mutazioni in SUFU3. Gli individui portatori di una mutazione in uno di questi due geni sono particolarmente sensibili all'esposizione al sole e possono sviluppare migliaia di tumori basocellulari nell'area cutanea esposta. Il rischio di sviluppare un BCC nel corso della vita nei soggetti portatori di una mutazione in PTCH1 è del 90%. I farmaci che bloccano la via difettosa causata dalle mutazioni in PTCH1 sono disponibili e possono essere prescritti per il cancro che si è diffuso o che non può essere trattato con la chirurgia o la radioterapia.
La sindrome di DICER1, causata da mutazioni nel gene DICER1, è una condizione rara che predispone a vari tumori benigni e maligni che si verificano principalmente nei bambini e negli adolescenti e danneggiano diversi organi come polmoni, reni, tiroide, ovaie e diverse altre sedi del corpo: o Il blastoma pleuropolmonare (PPB), un tumore polmonare cistico che può trasformarsi in un tumore polmonare invasivo, rappresenta la manifestazione più pericolosa. o Il gozzo multinodulare (MNG) è una patologia benigna della tiroide e, meno comunemente, il cancro della tiroide. o Tumori ovarici (tumore delle cellule di Sertoli-Leydig, sarcoma e ginandroblastoma). o Il nefroma cistico, un tumore benigno del rene, e, meno comune, il tumore di Wilms. o Rabdomiosarcoma embrionale della cervice uterina (e, meno comunemente, di altre sedi).
Il retinoblastoma è un tumore oculare che si sviluppa dalle cellule della retina e colpisce quasi esclusivamente i bambini di età inferiore ai 4-5 anni. I retinoblastomi esistono in forma ereditaria e sporadica. In entrambi i casi è coinvolta una mutazione del gene Rb1. La malattia può presentarsi unilaterale o bilaterale. Nel 60%-80% dei casi in cui la malattia si presenta su entrambi gli occhi e nel 15% dei casi di presentazione unilaterale, è stata rilevata una mutazione germinale in Rb1.
Il complesso di Carney (CNC) è una condizione autosomica dominante molto rara causata da mutazioni in PRKAR1A, che porta ad anomalie della pigmentazione cutanea. Le alterazioni endocrine possono manifestarsi con acromegalia, tumori tiroidei e testicolari e sindrome di Cushing indipendente dall'ormone adrenocorticotropo (ACTH), dovuta alla malattia adrenocorticale nodulare pigmentata primaria (PPNAD). Inoltre, i mixomi possono svilupparsi nel cuore, nella pelle e nel seno. I mixomi cardiaci possono svilupparsi in qualsiasi camera cardiaca, possono essere multipli e possono richiedere la rimozione chirurgica.
Il tumore rabdoide (RT) è un sarcoma pediatrico aggressivo dei tessuti molli che insorge nel rene, nel fegato, nel sistema nervoso centrale, nei nervi periferici e in tutte le varie parti molli del corpo. Il RT è causato dall'inattivazione biallelica di SMARCB1 (90% di tutti i casi) o SMARCA4 e nel 25% dei casi è associato a mutazioni germinali. In un caso è stata identificata una mutazione germinale in FBXW7, gene soppressore del tumore. La RT si manifesta solitamente nell'infanzia o nella fanciullezza, colpendo per lo più pazienti di età inferiore ai 2 anni. Nei bambini di età inferiore a un anno, la RT rappresenta circa il 20% dei tumori renali e il 15% dei tumori delle parti molli.
La sindrome di Von Hippel-Lindau (VHL) è una condizione ereditaria associata a tumori che insorgono in più organi, causati dalla mutazione del gene VHL. Nei pazienti con mutazione VHL si sviluppano emangioblastomi (tumori dei vasi sanguigni) nel cervello, nel midollo spinale e nella retina. Le persone affette da VHL hanno anche un rischio maggiore di sviluppare il carcinoma renale a cellule chiare (ccRCC), il tumore neuroendocrino del pancreas (pNET) e i tumori della ghiandola surrenale o il feocromocitoma.
Il nefroblastoma, o tumore di Wilms, è il più comune tumore maligno del rene nei bambini ed è associato a una proliferazione anomala di cellule embrionali simili al rene. Il tumore si sviluppa più frequentemente in un solo rene (unilaterale), ma in alcuni casi può colpire entrambi i reni (bilaterale). Nel 90% dei casi ha un'insorgenza sporadica e solo in una piccolissima percentuale di casi può essere ereditario. In almeno il 20% dei casi ereditari il tumore di Wilms è causato da mutazioni nel gene WT1. In alcuni casi è stata identificata una mutazione germinale in FBXW7, un gene soppressore tumorale frequentemente mutato nei tumori umani.
Mutazioni germinali nel gene soppressore del tumore FLCN sono responsabili della sindrome di Birt-Hogg-Dubé (BHD), una malattia ereditaria autosomica dominante che predispone a fibrofolliculomi, cisti polmonari che spesso causano pneumotorace spontaneo e un aumentato rischio di sviluppare tumori renali.
Il carcinoma a cellule renali papillari ereditario (HPRCC) è una sindrome familiare di cancro renale caratterizzata da una predisposizione allo sviluppo bilaterale e multifocale di carcinomi a cellule renali papillari di tipo 1. La predisposizione è trasmessa come carattere autosomico dominante con penetranza ridotta. La predisposizione è trasmessa come carattere autosomico dominante con penetranza ridotta. La sindrome è associata a mutazioni germinali del protooncogene MET.
La leiomiomatosi ereditaria e a cellule renali (HLRCC) consiste nella presenza di leiomi multipli cutanei in diversi individui della stessa famiglia. I leiomiomi sono neoplasie benigne dei tessuti molli che hanno origine dalla muscolatura liscia. Questa rara sindrome è solitamente trasmessa in modo autosomico dominante e può essere associata a tumori come i leiomiomi uterini e il carcinoma renale. La malattia è causata da mutazioni germinali nel gene della fumarato idratasi FH.
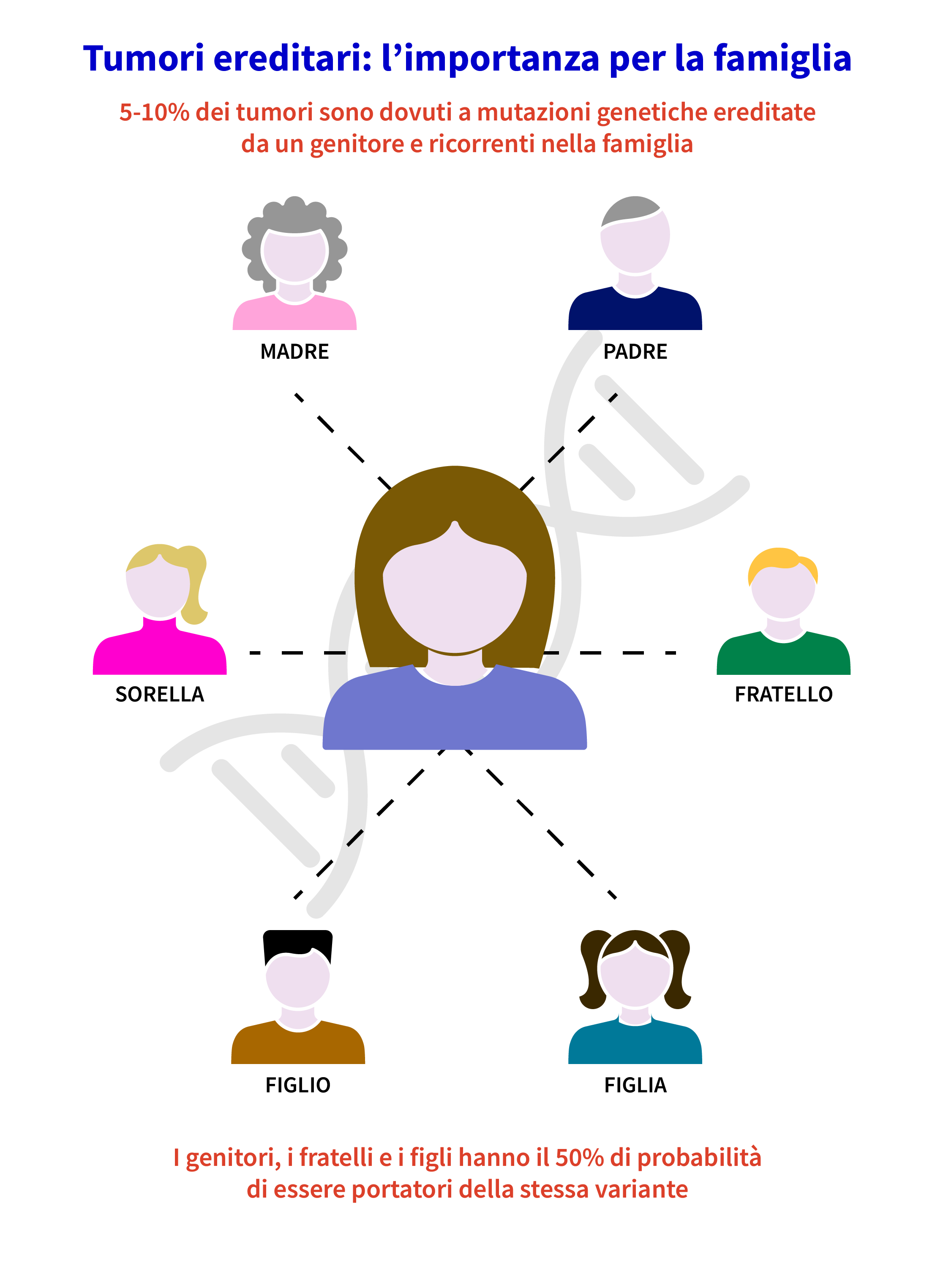
Analisi delle mutazioni nei parenti
Una volta identificata una mutazione in un probando, è importante offrire il test ai familiari, per fornire opzioni più appropriate in termini di screening preventivo, terapia e chirurgia, sulla base del rischio personale e in linea con le scelte personali.
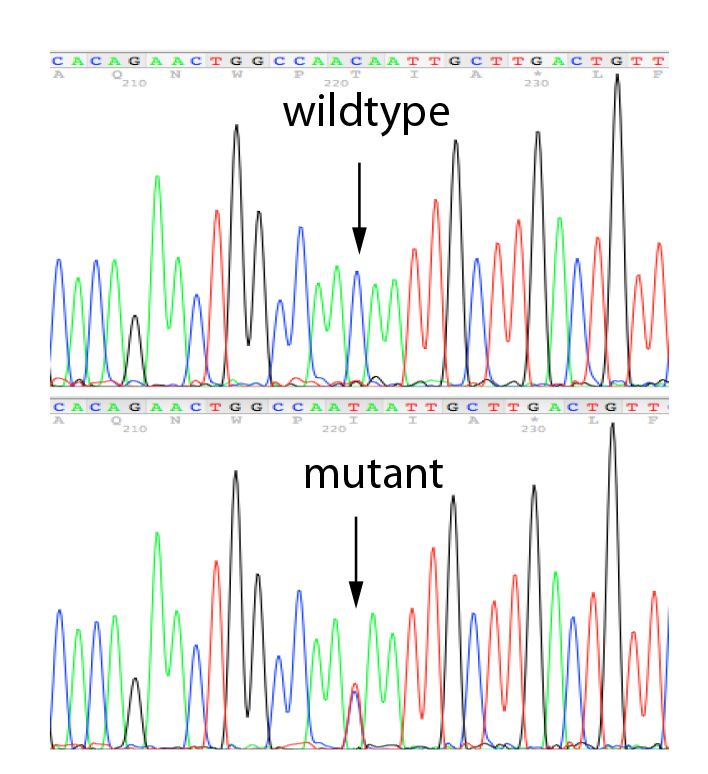
A seconda del tipo di variante identificata nel probando, il test sui familiari sarà effettuato con il sequenziamento Sanger (per verificare la presenza di una variante puntiforme) o con l'MLPA (per identificare grandi delezioni o amplificazioni di un gene). La tecnica MS-MLPA può essere utilizzata per valutare lo stato di metilazione del gene MLH1, correlato alla sindrome di Lynch.
Il sequenziamento Sanger è una strategia di analisi robusta in grado di determinare la presenza di una mutazione puntiforme o di una piccola delezione/duplicazione. È ampiamente utilizzato per identificare varianti costituzionali (germinali) nei test diagnostici attraverso la lettura di una piccola regione target del genoma. Effettuiamo il sequenziamento Sanger utilizzando i nostri set di primer convalidati per la convalida delle varianti identificate in uno qualsiasi dei geni presenti nei nostri pannelli NGS e per l'analisi dei parenti.
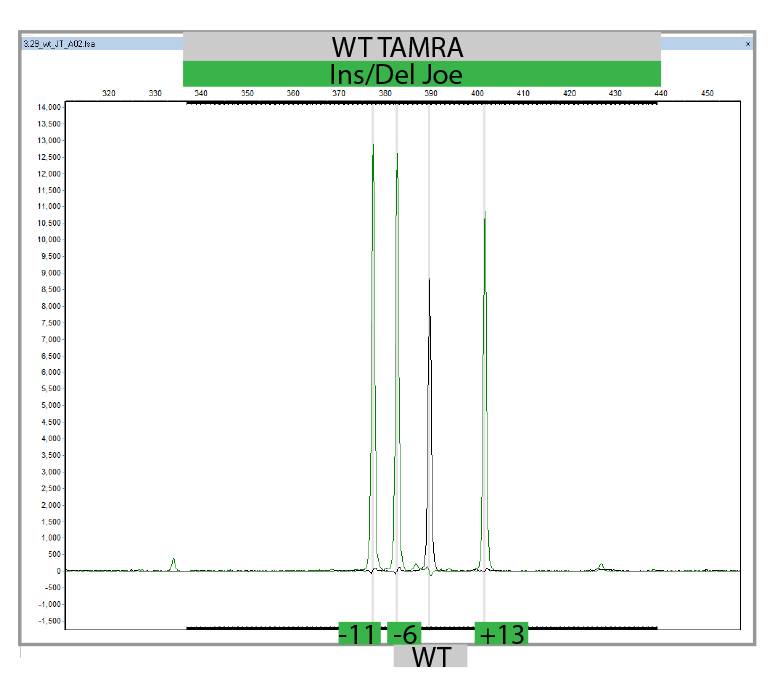
L'MLPA è una semplice tecnica di PCR multipla che utilizza una singola coppia di primer per amplificare fino a 60 sonde, ciascuna con un target genomico e una lunghezza unici. Gli ampliconi della PCR sono marcati in modo fluorescente e separati e quantificati mediante elettroforesi capillare. Confrontando il pattern di picchi risultante di un campione con quello di una serie di campioni di riferimento, è possibile determinare il numero di target genomici presenti nel campione di interesse. L'analisi MLPA può anche essere utilizzata per determinare se una specifica regione genomica è metilata (MS-MLPA), il che influenza l'espressione di determinati geni. Eseguiamo l'MLPA per la convalida di risultati identificati in uno qualsiasi dei geni presenti nei nostri pannelli NGS di proprietà, utilizzando i kit Salsa forniti da MRC-Holland e per l'analisi dei parenti.
Clicca qui per la scheda tecnica dell'MLPA.
Clicca qui per la scheda tecnica dell'MS-MLPA.
Terapia personalizzata
Il pannello OncoPan®, oltre a identificare varianti che aumentano il rischio di cancro, è anche progettato per aiutare a indirizzare la terapia con gli inibitori di PARP (PARPi):
- Test BRCA1/2 somatico per la terapia PARPi nel cancro al seno, all'ovaio, alla prostata e al pancreas
- Test OncoHRD per la terapia con PARPi nel carcinoma ovarico (secondo le linee guida europee)
La terapia con inibitori di PARP si basa sul fatto che una cellula normale ha due modi di riparare i danni al DNA: La riparazione della rottura del DNA a singolo filamento dipendente da PARP e la riparazione della rottura del DNA a doppio filamento dipendente da BRCA1/2. In caso di rottura del DNA a singolo filamento, la proteina PARP si lega al DNA e recluta altre proteine che riparano il danno. In presenza di inibitori della PARP, quest'ultima si lega ancora al DNA ma non può più reclutare le proteine di riparazione. Di conseguenza, il danno non viene riparato e, peggio ancora, si accumula altro danno al DNA sotto forma di rotture del DNA a doppio filamento. In una cellula normale, la riparazione delle rotture del DNA a doppio filamento dipendente da BRCA1/2 elimina il danno e la cellula si riprende. In una cellula cancerosa che presenta una mutazione in BRCA1/, le rotture del DNA a doppio filamento non possono più essere riparate e il danno al DNA si accumula fino a quando la cellula non può più funzionare e muore.
Una mutazione BRCA1/2 in un tumore può essere germinale o somatica. Se prima della malattia non è stato valutato lo stato di mutazione germinale, verrà eseguito un test somatico, che rivelerà sia le mutazioni germinali che quelle somatiche, per valutare l'idoneità al trattamento con la terapia con PARP inibitore.
Il test somatico prevede la ricerca di mutazioni in BRCA1/2 e potrebbe essere esteso ad altri geni coinvolti nel meccanismo di riparazione del DNA a doppio filamento dipendente da BRCA1/2 per valutare il cosiddetto deficit di ricombinazione omologa o HRD. In presenza di HRD, si crea un modello di mutazione specifico nel DNA (cicatrice HRD) che può essere identificato mediante il sequenziamento dell'intero genoma.
L'HRD è presente in molti tumori diversi. Ad esempio, il 50% dei tumori ovarici epiteliali presenta HRD, ma solo il 15% ospita mutazioni BRCA1/2 germinali o somatiche (Konstantinopoulos et al. Cancer Discov 2015;5:1137-54.). Un numero simile si riscontra nei tumori al seno tripli negativi, con il 15% che ospita mutazioni BRCA1/2 germinali, mentre un ulteriore 40% di tumori presenta una cicatrice HRD dovuta a mutazioni BRCA1/2 somatiche o a mutazioni in altri geni della via (Grace et al., Breast Cancer Research and Treatment 2022, 192, 649-653). Nei tumori della prostata in fase avanzata fino al 25% dei tumori presenta una cicatrice HRD. (Handy et al., 2018, JCO, 36.15_suppl, 5062).
Attualmente, secondo le linee guida europee, la terapia con PARP inibitori è raccomandata come terapia di mantenimento per il carcinoma ovarico, il carcinoma mammario HER2 negativo e il carcinoma prostatico (per i dettagli, EMEA e ESMO ). I pazienti eleggibili vengono identificati mediante screening mutazionale in BRCA1/2 o mediante test per HRD (tumori ovarici). Cogentech offre sia lo screening per le mutazioni BRCA1/2 sia l'analisi per l'HRD utilizzando il proprio pannello OncoPan®.
L'analisi mutazionale di BRCA1/2 mediante OncoPan® viene eseguita su campioni tumorali FFPE per identificare i pazienti che potrebbero essere idonei alla terapia con PARPi.
Questo test viene offerto a scopo di ricerca dal laboratorio di Milano.
Il test OncoHRD combina l'analisi mutazionale di BRCA1/2 mediante OncoPan® con un sequenziamento a basso passaggio dell'intero genoma (lp-WGS) per verificare la presenza di difetto del sistema di ricombinazione omologa (Homologous Recombination Deficiency o HRD) utilizzando lo strumento bioinformatico GIInger™ di Sophia Genetics per identificare i pazienti che potrebbero essere idonei alla terapia con PARPi.
Questo test viene offerto a scopo di ricerca dal laboratorio di Milano.
Clicca qui per la scheda tecnica di OncoHRD.
